
Rare Disease Day, observed annually on the last day of February, raises awareness for the millions of people worldwide living with rare and often poorly understood conditions. Two recently published studies shed light on the neurological mechanisms underlying Timothy syndrome and Alexander disease. Despite their distinct causes, both rare disorders share a common thread in how genetic mutations disrupt brain function at the synaptic and circuit level. These studies employ ex–vivo brain slice electrophysiology as a key tool, using precisely sectioned tissue to bridge the gap between molecular pathology and functional neural circuit deficits. In tandem, they demonstrate how this approach can effectively reveal shared physiological vulnerabilities across vastly different rare disease models.
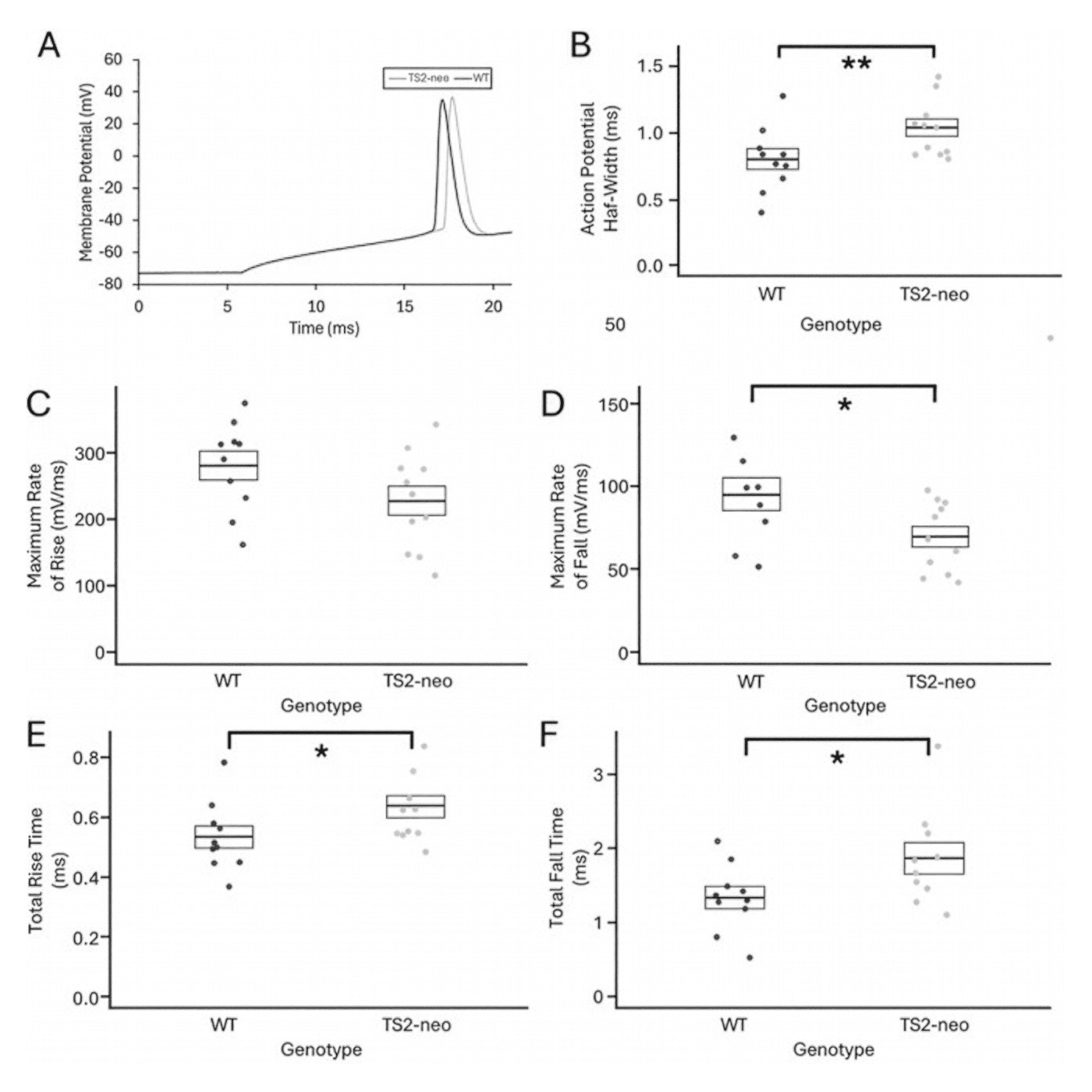
Note: Featured image from Craddock et al., (2025) (licensed under CC BY 4.0) showing how the TS2-neo mutation causes an increase in action potential (AP) width. A) Representative AP waveforms from V1 pyramidal cells of a TS2-neo mouse (gray) and a WT mouse (black), B) AP half-widths in V1 pyramidal cells from WT (left, n = 10) and TS2-neo (right, n = 11) mice, C) Maximum rates of AP rise and D) fall, and E) total rise time and F) total fall time in V1 pyramidal cells in WT and TS2-neo mice (* indicates significant difference p < 0.05 which was lost after correction for multiple comparisons, and ** indicates significant difference p < 0.05 both before and after correction for multiple comparisons).
Disruptions in primary visual cortex physiology and function in a mouse model of Timothy syndrome
Timothy syndrome (TS) is a rare genetic disorder caused by mutations in the CACNA1C gene encoding the CaV1.2 L-type calcium channel. It is associated with serious cardiac and developmental symptoms, as well as neuropsychiatric features, including autism and atypical sensory processing. Although prior studies in patient-derived neurons have identified cellular and electrophysiological abnormalities, the effects of TS mutations on functional cortical circuits within sensory systems remain poorly understood. Berman et al (2025). addressed that gap by using a TS2-neo mouse model to investigate how the TS mutation alters neuronal physiology and visual processing in the primary visual cortex (V1).
The group used heterozygous TS2-neo mice to investigate visual cortex function through ex-vivo electrophysiology, in-vivo two-photon calcium imaging, and immunohistochemistry. For slice electrophysiology, 400 μm coronal brain sections containing V1 were prepared using a Campden Instruments 7000smz-2 vibrating microtome. This enabled precise tissue sectioning prior to whole-cell patch-clamp recordings of pyramidal neurons to measure membrane and action potential properties.
Ex-vivo whole-cell recordings from V1 pyramidal neurons showed that although passive membrane properties and AP threshold and amplitude were unchanged, TS2-neo mice exhibited a significant prolongation of action potential half-width, consistent with effects of classical TS mutations in other excitable cells. In-vivo two-photon calcium imaging demonstrated that TS2-neo heterozygosity significantly shifted neuronal contrast sensitivity functions, with reduced sensitivity to low spatial frequency stimuli and relatively greater responsiveness at medium-to-high spatial frequencies. TS2-neo mice also showed an increase in parvalbumin-positive interneuron density in V1. Collectively, these findings indicate that classical TS mutations alter spike dynamics, inhibitory circuit composition, and sensory processing in the visual cortex, providing circuit-level insight into how CACNA1C dysfunction may contribute to sensory abnormalities associated with Timothy syndrome and related autism spectrum phenotypes.
Gfap Mutation and Astrocyte Dysfunction Lead to a Neurodegenerative Profile with Impaired Synaptic Plasticity and Cognitive Deficits in a Rat Model of Alexander Disease
Alexander disease (AxD) is a progressive, fatal central nervous system disorder for which 90% of cases are caused by missense mutations in the gene encoding glial fibrillary acidic protein (GFAP). This leads to astrocyte dysfunction, Rosenthal fiber formation, and white matter deficits with a range of cognitive and motor impairments. Although AxD was originally described as an intellectual disability syndrome, cognitive dysfunction has received little systematic attention. Key questions, including which cognitive domains are affected, how impairments relate to known sites of pathology, and whether they can serve as outcome measures for experimental treatments remain unanswered. Craddock et al. (2025) leveraged a newly developed rat model of AxD that more closely recapitulates the human disease than earlier mouse models to investigate how GFAP mutations in astrocytes impair synaptic, neuronal, and cognitive functions.
The study used a Gfap+/R237H rat model of AxD to investigate whether astrocyte-induced impairments were seen in cognitive functioning. Hippocampal tissue was used for RNA sequencing, western blotting, and cytokine assays, while neuronal populations were quantified via stereology. Synaptic function was assessed through LTP recordings in 400 µm coronal slices prepared with a Campden Instruments 7000smz-2 vibrating microtome. Cognitive function was then evaluated behaviorally, using the novel object recognition test and Barnes maze tasks.
Hippocampal transcriptomics revealed upregulated immune-related genes and downregulated synaptic and mitochondrial transcripts, with the neurodegenerative profile worsening as disease progressed. Protein analysis confirmed reductions in synaptic markers and mitochondrial complex proteins, while stereology identified selective granule cell loss in the dentate gyrus. LTP recordings demonstrated impaired synaptic plasticity in R237H rats, and behavioral testing confirmed deficits in novel object recognition and spatial memory. Together, this established neuronal and cognitive impairment as clinically relevant phenotypes in the rat model of AxD and highlighted the broader role of astrocytes.
Conclusions
These studies by Craddock et al. (2025) and Berman et al. (2025). advance our understanding of how genetic mutations disrupt neural circuit function in rare neurological disorders. In effect, they demonstrate how both CaV1.2 dysfunction in Timothy syndrome and GFAP mutation in Alexander disease produce disruptive changes at the synaptic, cellular, and behavioral levels. On this Rare Disease Day, these findings serve as a reminder that investigating even the rarest conditions can illuminate fundamental mechanisms of brain function and neurodegeneration, with implications far beyond the diseases themselves.


Upcoming Events
-
Oxford Neuroscience Symposium
Date: March 18, 2026 -
Cambridge Neuroscience Seminar
Date: April 1, 2026 -
Symposium of the Young Physiologists
Date: April 8, 2026
